
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly spread across the world over the last two years. With substantial human reservoirs with limited immunity, many new SARS-CoV-2 variants and sublineages of these variants have arisen.
 Study: SARS-CoV-2 within-host and in-vitro genomic variability and sub-genomic RNA levels indicate differences in viral expression between clinical and in-vitro cohorts.
Study: SARS-CoV-2 within-host and in-vitro genomic variability and sub-genomic RNA levels indicate differences in viral expression between clinical and in-vitro cohorts.
Even individuals who have contracted coronavirus disease 2019 (COVID-19) previously, or have been vaccinated, are still at risk, as reports of reinfections and vaccine breakthrough infections increase daily. SARS-CoV-2 strains such as the Delta and Beta variants show significant mutations in the spike protein that can help them evade immune responses. In a recent study published on the preprint server medRxiv*, researchers from the University of Sydney investigate the genomic variability of SARS-CoV-2 within the host and in vitro.
About the study
In the current study, the researchers collected specimens five to 23 days after COVID-19 symptoms began or after the first positive test. A total of 90 specimens were gathered from individuals who had received a positive reverse-transcriptase qualitative polymerase chain reaction (RT-qPCR) test.
The collected samples included nasopharyngeal swabs, as well as lower respiratory tract samples such as bronchoalveolar lavages and sputum. The samples were split between those that had been gathered from patients with severe disease, which was classified as requiring intubation or intensive care unit (ICU) admission, and mild cases that did not require care. Samples that tested negative for SARS-CoV-2 were used as negative controls.
The scientists conducted daily sampling to discoverer the 50% tissue culture infective dose (TCID50) of SARS-CoV-2 from the wild-type strain, as well as the Beta and Delta variants. Samples were sequenced to confirm lineage. The raw sequences were analyzed both in-house and later in downstream analysis.
Only genomes that received over 80% coverage over a 100x depth in all variant positions were considered in the final analysis. The Mann-Whitney test, which determines the difference between means on variables, was used to analyze the intrahost single nucleotide variant (iSNV)/ single nucleotide polymorphism (SNP) counts, read frequencies, and single guide ribonucleic acid (sgRNA) counts.
Study findings
After genome and variant level filtering, as well as quality control, was performed, a total of 25 SARS-CoV-2 genomes were recovered from the mild cases and 26 genomes from the severe cases. Surprisingly, the researchers found no difference between the two cohorts in age and sex, despite multiple studies showing strong links between both factors and more severe disease in men and the elderly. However, the sample size in this study was much smaller than those studies.
There was also no significant difference in viral load between cohorts. A wide range of sublineages was detected from the samples, the majority of which originated from either the Delta or Beta strains.
Low-frequency iSNVs were detected in all severe cases and 83% of the mild cases. The difference in numbers of iSNVs per SARS-CoV-2 gene between the two cohorts was only significant for iSNVs that affected the spike gene, while the only significant difference for total counts of iSNVs was seen in the ORF1ab gene.
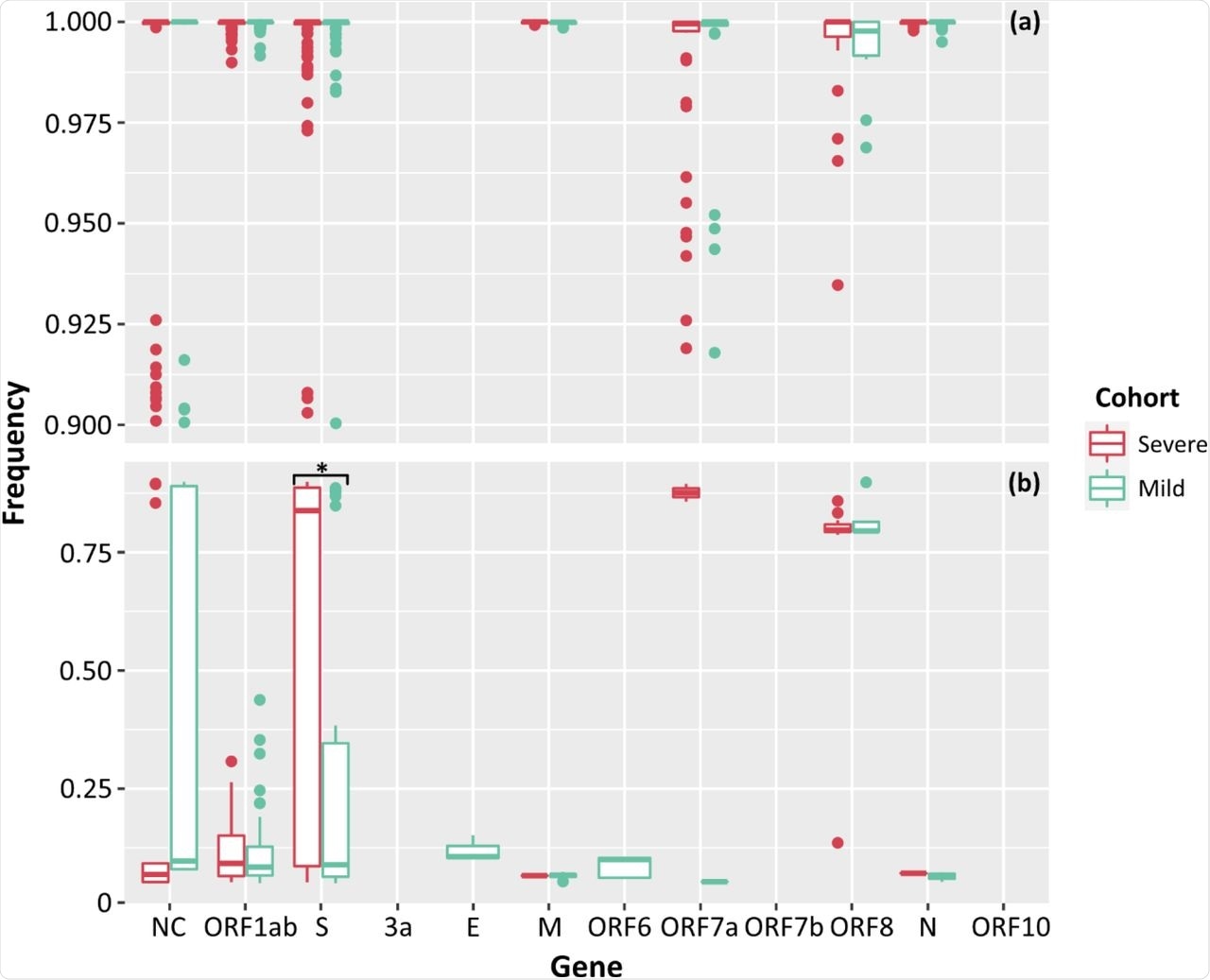 Frequencies of SNPs (a) and iSNVs (b) by SARS-CoV-2 gene for severe (red), and mild (green) cohorts. Frequencies ≥ 0.9 were considered SNPs. Problematic sites are not included. NC signifies non-coding region of the genome. Statistical significance (p ≤ 0.05) is denoted by (*). The frequency of iSNVs in the spike gene of SARS-CoV-2 were significantly different between the severe and mild cohorts.
Frequencies of SNPs (a) and iSNVs (b) by SARS-CoV-2 gene for severe (red), and mild (green) cohorts. Frequencies ≥ 0.9 were considered SNPs. Problematic sites are not included. NC signifies non-coding region of the genome. Statistical significance (p ≤ 0.05) is denoted by (*). The frequency of iSNVs in the spike gene of SARS-CoV-2 were significantly different between the severe and mild cohorts.
The researchers found four severe cases of especial interest. The SNP profile of each of these patients remained comparatively fixed over time, with a general trend showing an increase in the number of iSNVs over the course of the earlier lineage infections.
Case P0105 displayed a single iSNV that remained consistent across three samples from seven to ten days post-symptom onset. This iSNV was ultimately lost at 11 days; however, the sample showed iSNVs at eight new locations within the genome.
Case P0322 showed no consistent iSNVs; however, an average of three iSNVs was found per sample. On day nine, iSNVs were present at 12 positions and three positions on day ten. In samples showing the B.1.617.2 sublineages, iSNVs tended to be lost rather than gained, and there were fewer conserved iSNVs.
For case P608, one iSNV and one low-frequency deletion event remained present for a full 11 days, and one iSNV present at day one converted to an SNP on day 11. A high-frequency deletion event converted to a low-frequency deletion event on the same date.
In the case of P0615, one iSNV and two low-frequency deletion events were retained from day three to day eight, with one iSNV being lost at day eight.
Conclusions
The authors highlight that their study shows the dynamic difference in the position and frequencies of iSNVs in both mild and severe cohorts over time. They urge continual monitoring, especially in community outbreaks where multiple variants may be circulating at the same time. This information could be invaluable for epidemiologists and geneticists attempting to track the progress and future evolution of the disease.
*Important notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information
- Agius, J. E., Johnson-Mackinnon, J. C., Fong, W., et al. (2021). SARS-CoV-2 within-host and in-vitro genomic variability and sub-genomic RNA levels indicate differences in viral expression between clinical and in-vitro cohorts. medRxiv. doi:10.1101/2021.11.23.21266789. https://www.medrxiv.org/content/10.1101/2021.11.23.21266789v1
Posted in: Genomics | Medical Science News | Medical Research News | Disease/Infection News
Tags: Coding Region, Coronavirus, Coronavirus Disease COVID-19, Evolution, Frequency, Gene, Genome, Genomic, immunity, in vitro, Intensive Care, Nasopharyngeal, Nucleotide, Polymerase, Polymerase Chain Reaction, Protein, Respiratory, Ribonucleic Acid, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Single Nucleotide Polymorphism, Spike Protein, Syndrome, Tissue Culture, Vaccine

Written by
Sam Hancock
Sam completed his MSci in Genetics at the University of Nottingham in 2019, fuelled initially by an interest in genetic ageing. As part of his degree, he also investigated the role of rnh genes in originless replication in archaea.
Source: Read Full Article