
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is responsible for the coronavirus disease 2019 (COVID-19), has caused almost 4.5 million deaths worldwide and infected almost 211 million as of August 19, 2021.
To date, millions of doses of the COVID-19 vaccines have been deployed globally, which has significantly reduced the rate of infections, hospitalizations, and deaths from COVID-19. Despite these efforts, emerging SARS-CoV-2 variants that evade neutralization by antibodies to earlier lineages have threatened the efficacy of current vaccines.
A new study published on the preprint server bioRxiv* explores the mechanism by which the SARS-CoV-2 lambda variant has acquired immune evasion capabilities above that of the wildtype. This appears to involve shortening of the epitopes on the viral spike protein, as well as acquiring additional sites that can be N-glycosylated.
 Study: Cutting epitopes to survive: the case of lambda variant. Image Credit: manaemedia / Shutterstock.com
Study: Cutting epitopes to survive: the case of lambda variant. Image Credit: manaemedia / Shutterstock.com
Background
The SARS-CoV-2 lambda variant was first identified in Peru, but has quickly spread to other parts of South America and the United States. The spike protein of SARS-CoV-2, which contains the viral receptor-binding domain (RBD), binds to the host cell angiotensin-converting enzyme 2 (ACE2) receptor.
Although data has demonstrated how the RBD of SARS-CoV-2 mediates the entry of this virus into the cell, its precise interaction with the host cell is not fully understood. Mutations in both the spike protein and the RBD are key to the heightened resistance of new SARS-CoV-2 variants to antibody-mediated neutralization and infectivity.
About the study
In the current study, the scientists compared the spike genetic sequence of the lambda variant and the reference Wuhan spike sequence. The mutations observed in the RBD were modeled on the crystal structure using appropriate software.
After incorporating the point mutations observed in this variant, their effect on the spike structure and glycosylation sites was predicted by computational modeling.
Study findings
The results showed that two of the lambda mutations, G75V and T76I, were located at an exposed loop between two antiparallel β-strands. Both of these mutations were found to stabilize the spike glycoprotein. Additionally, these two mutations appear to ensure that the loop remains in contact with another loop between positions 246-280, which is a binding site for monoclonal antibodies (MAbs) specific to the virus.
The deletion mutation of 246-252 is also located in this loop. This deletion may affect the affinity of binding of the N-terminal domain (NTD) of the spike to human MAbs.
To confirm the role of this deletion mutation on the NTD, a spike-4A8 Mab complex was studied in detail. The mutant NTD was superimposed on the wildtype domain of the spike-antibody complex.
Taken together, this in silico experiment showed that the MAb interacts less strongly with the loop when the partial deletion is present, thus causing weaker binding affinity as a result of the loss of several potential interactions. This key NTD deletion in the lambda strain results in the loss of interactions between L249 and both F60 and Y54 of the 4A8 MAb light chain.
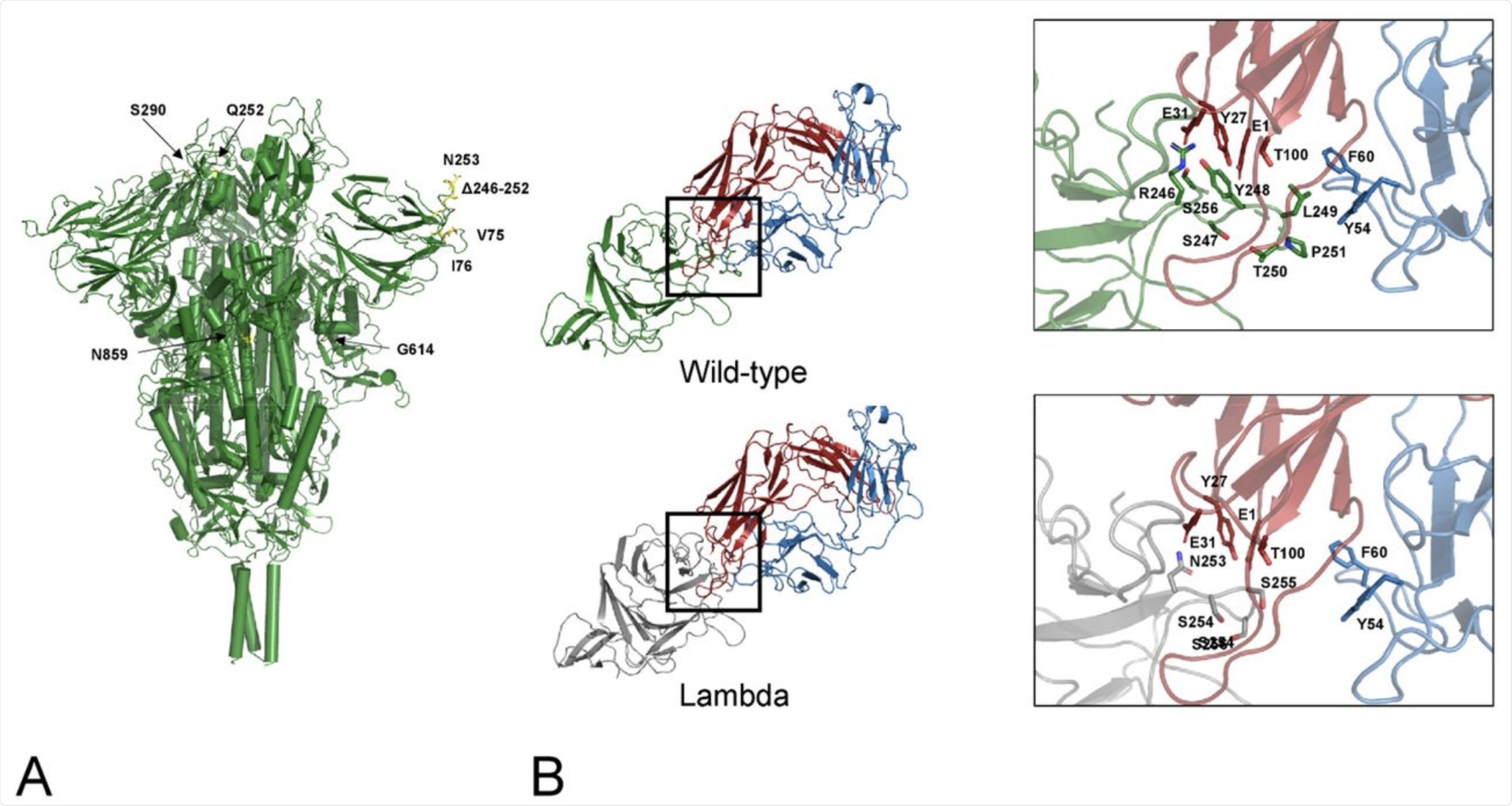 A) Map of the lambda mutations onto the trimeric Spike structure. Only one mutation per monomer has been indicated. B) interface NTD-4A8. The mutant NTD has been modelled via SwissModel and superposed to the wild-type domain of the complex. Interface interactions and energies calculated by PRODIGY have been compared. Comparison of the interfaces between the wild-type (green cartoon) and the lambda NTD (grey cartoon) and the mAb 48A at the deletion region. Red and blue cartoons indicate the heavy and light chain respectively. Side chains in the deleted loop and the interacting residues are indicated with stick models and labelled. Sequence numbering refers to the wild-type Spike.
A) Map of the lambda mutations onto the trimeric Spike structure. Only one mutation per monomer has been indicated. B) interface NTD-4A8. The mutant NTD has been modelled via SwissModel and superposed to the wild-type domain of the complex. Interface interactions and energies calculated by PRODIGY have been compared. Comparison of the interfaces between the wild-type (green cartoon) and the lambda NTD (grey cartoon) and the mAb 48A at the deletion region. Red and blue cartoons indicate the heavy and light chain respectively. Side chains in the deleted loop and the interacting residues are indicated with stick models and labelled. Sequence numbering refers to the wild-type Spike.
Other mutations
The L452Q and F400S point mutations in the RBD of the SARS-CoV-2 lambda variant do not appear to significantly affect the stability of the domain; however, these mutations do convert the hydrophobic residue here into a hydrophilic one. The result is a highly hydrophilic surface of the RBD.
These sites are susceptible to solvation near the RBD-ACE2 junction, with both the wildtype and mutant RBD showing nearly the same binding affinity. The S1 domain’s D614G mutation arose early and was responsible for the rapid rise of the lineage to global dominance.
In the S2 subunit of the spike protein, the T859N is expressed near the G614 mutant of the neighboring subunit. Overall, this mutation has not been predicted to affect the stability of the molecule.
Interestingly, as a result of this change, a new hydrogen bond to H487 occurs. The researchers suggest that the two mutations of D614G and T859N can together trigger a long-range allosteric effect that impacts the structural flexibility of the complex
Implications
This prototype modeling analysis of the RBD-receptor complex seems to dissociate the mutations that define the lambda variant from any effect on the RBD-ACE2 binding affinity. The mechanism by which the former affects the transmission of the virus is still uncertain.
One explanation is that the S2 domain mutations exert an allosteric effect at long-range, which changes the flexibility of the protein’s conformation and its affinity for the ACE2 receptor. A better suggestion is that the 246-252 deletion in the lambda variant is likely responsible for its functional change that allows this variant to escape the host immune response.
The ability of the lambda variant to escape the immune response is likely achieved by two possible routes. One explanation is the shortening of antibody binding sites on the loops of the spike protein, while the other is by changing the level of glycosylation upwards. These factors have already been found to occur during viral transmission protect the virus from antibody recognition.
In another recent preprint study, a seven-residue deletion at the NTD of the lambda variant was observed. This deletion could make this variant resistant to antiviral immunity, thus agreeing with the findings of the current study.
“Biophysical and bioinformatics data suggest that a combination of shortening immunogenic epitope loops and generation of potential N-glycosylation sites may be a viable adaptation strategy potentially allowing this emerging viral variant escaping host immunity.”
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Pascarella, S., Ciccozzi, M., Bianchi, M., et al. (2021). Cutting epitopes to survive: the case of lambda variant. bioRxiv. doi:10.1101/2021.08.14.456353. https://www.biorxiv.org/content/10.1101/2021.08.14.456353v1.
Posted in: Molecular & Structural Biology | Genomics | Medical Science News | Life Sciences News | Medical Research News | Medical Condition News | Disease/Infection News | Healthcare News
Tags: ACE2, Angiotensin, Angiotensin-Converting Enzyme 2, Antibodies, Antibody, binding affinity, Bioinformatics, Cell, Computational Modeling, Coronavirus, Coronavirus Disease COVID-19, Efficacy, Enzyme, Genetic, Glycoprotein, Glycosylation, Immune Response, immunity, Molecule, Mutation, Protein, Receptor, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome, Virus

Written by
Dr. Liji Thomas
Dr. Liji Thomas is an OB-GYN, who graduated from the Government Medical College, University of Calicut, Kerala, in 2001. Liji practiced as a full-time consultant in obstetrics/gynecology in a private hospital for a few years following her graduation. She has counseled hundreds of patients facing issues from pregnancy-related problems and infertility, and has been in charge of over 2,000 deliveries, striving always to achieve a normal delivery rather than operative.
Source: Read Full Article